General Information About Gastrointestinal Stromal Tumors (GIST)
Epidemiology
Although they comprise fewer than 1% of all gastrointestinal (GI) tumors, GIST are the most common mesenchymal tumors of the GI tract.[1] It has been estimated that there are 3,300 to 6,000 new GIST cases per year in the United States.[2] A study based on Surveillance, Epidemiology and End Results (SEER) registry data found that the age-adjusted yearly incidence of GIST in the United States was 6.8 per million from 1992 to 2000.[3] However, the true incidence is not known, in part because many tumors have not been tested for the characteristic KIT or platelet-derived growth factor receptor alpha (PDGFRA) gene mutations. In addition, small, indolent GIST, only a few millimeters in diameter, are common in the general population and are not included in cancer registries.[4,5] GIST are equally distributed across all geographic and ethnic groups and men and women are equally affected. Most patients present between the ages of 50 and 80.[6] The vast majority of GIST are sporadic, but there are rare familial forms associated with the characteristic heritable mutations in the KIT gene (or, rarely, in succinate dehydrogenase genes in Carney-Stratakis syndrome). Familial GIST may present as multiple primary tumors.
Clinical Presentation and Diagnostic Evaluation
GIST can occur anywhere along the GI tract, but most often are found in the stomach or small intestine. The American Joint Committee on Cancer (AJCC) Cancer Staging Manual lists the following approximate distributions:[7]
- Stomach (60%).
- Small intestine (30%).
- Rectum (3%).
- Colon (1–2%).
- Esophagus (<1%).
- Omentum/mesentery (rare).
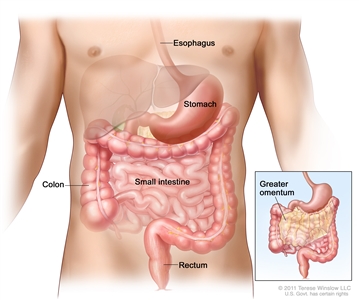
Gastrointestinal stromal tumors (GISTs) may be found anywhere in or near the gastrointestinal tract.
Less frequently, GIST may arise in the appendix, gallbladder, pancreas, retroperitoneum, and paravaginal and periprostatic tissues.[8] Approximately 20% to 25% of gastric GIST and 40% to 50% of small intestinal GIST are clinically aggressive.[9,10] It has been estimated that approximately 10% to 25% of patients present with metastatic disease.[9,11]
The clinical presentation of patients with GIST varies depending on the anatomic location of the tumor and the tumor size and aggressiveness.[12] The most common presentation of GIST is GI bleeding, which may be acute (melena or hematemesis) or chronic and results in anemia.[10]
GIST patients may also present with:
- An acute abdomen caused by tumor rupture.
- GI obstruction.
- Appendicitis-like pain.
Other clinical symptoms include the following:[2]
- Fatigue.
- Dysphagia.
- Satiety.
Smaller lesions may be incidental findings during surgery, radiologic studies, or endoscopy. The natural history of these incidental tumors and the frequency of progression to symptomatic disease are unknown. There may be a substantial reservoir of small GIST tumors that do not progress to symptomatic stages. For example, a series of 98 consecutive systematic autopsies on adults who died of unrelated causes revealed grossly recognizable gastric tumors (1 mm–6 mm) that were histologically diagnosed as GIST in 22.5% of cases.[5] Sufficient DNA was available for analysis in 26 patients, revealing 13 patients with mutations in KIT exon 11 and one in PDGFRA.
In a retrospective study of 200 GIST cases, typical clinical manifestations of malignancy included liver metastases and/or dissemination within the abdominal cavity. Lymph node involvement and spread to the lungs or other extra-abdominal sites was unusual.[11] Advanced disease may be associated with metastases to distant sites, including lung and bone. Brain metastases are rare.[2]
GIST should be included in the differential diagnosis of any intra-abdominal nonepithelial malignancy. Diagnostic interventions may include the following:[12]
- Computed tomography (CT).
- Magnetic resonance imaging.
- Upper GI endoscopy.
Tests that may be useful in staging include the following:
- 18F-FDG PET (fluorine F 18-fludeoxyglucose positron emission tomography).
- CT.
Endoscopic ultrasound with fine-needle aspiration biopsy is useful in the detection of GIST in the upper GI tract because most tumors arise below the mucosal layer and grow in an endophytic fashion.[12,13,14]
Because nodal metastasis is so rare at diagnosis (i.e., it is virtually unheard of for true GIST according to the AJCC Cancer Staging Manual[7]), there is general agreement that nodal dissection is not needed.
Pathology and Molecular Genetics
Typically arising within the muscle wall of the GI tract, GIST range in size from less than 1 cm to more than 40 cm, with an average size of approximately 5 cm when diagnosed clinically.[2] Small GIST may form solid subserosal, intramural, or, less frequently, polypoid intraluminal masses. Large tumors tend to form external masses attached to the outer aspect of the gut involving the muscular layers.[2] GIST morphology is quite varied; the tumors are composed of the following:[8]
- Spindle cells (70%).
- Epithelioid cells (20%).
- Mixed spindle and epithelioid cells (10%).
GIST encompass a broad continuum of histologic patterns, ranging from bland-appearing tumors with very low mitotic activity (often previously designated leiomyomas) to very aggressive-appearing patterns (previously often called leiomyosarcomas).[7] They may originate from interstitial cells of Cajal (ICC) or their stem cell-like precursors, although this is not certain.[15,16]
The most commonly used marker for GIST is the CD117 antigen, a marker expressed by ICC. Approximately 95% of GISTs are positive for the CD117 antigen, an epitope of the KIT receptor tyrosine kinase.[2,9] However, CD117 immunohistochemistry is not specific for GIST, as weak reactivity occurs with other mesenchymal neoplasms; accordingly, morphologic examination and the use of other immunostains in difficult cases are indispensable.[17] In addition, false-positive CD117 staining can occur if antigen retrieval techniques are used in the pathology laboratory to enhance marker expression.[18] Because of a relatively broad morphologic spectrum, the differential diagnosis of GIST includes several mesenchymal, neural, and neuroendocrine neoplasms that occur in the abdomen including the following:[8]
- Leiomyoma.
- Leiomyosarcoma.
- Schwannoma.
- Malignant peripheral-nerve sheath tumor.
- Solitary fibrous tumor.
- Inflammatory myofibroblastic tumor.
- Fibromatosis.
- Synovial sarcoma.
- Neuroendocrine tumors (carcinoid and islet cell).
- Gastric glomus tumor.
- Malignant mesothelioma.
- Angiosarcoma.
- Sarcomatoid carcinoma.
Approximately 85% of GIST contain oncogenic mutations in one of two receptor tyrosine kinases: KIT or PDGFRA (platelet-derived growth factor receptor alpha).[2,10] Constitutive activation of either of these receptor tyrosine kinases plays a central role in the pathogenesis of GIST.[15,19] Wild-type tumors, with no detectable KIT or PDGFRA mutations, account for 12% to 15% of all GIST. Fewer than 5% of GIST occur in the setting of syndromic diseases, such as neurofibromatosis type 1 (NF1), Carney triad syndrome, and other familial diseases.[2,20,21,22] The correct identification of GIST is very important because of the availability of specific, molecular-targeted therapy with KIT/PDGFRA tyrosine kinase inhibitors (TKI) such as imatinib mesylate or, in the case of imatinib-resistant GIST, sunitinib malate.[1,10,17]
Risk Assessment and Prognosis
At the time of clinical presentation, the prognosis appears to be influenced by genetic events other than kinase mutations, although a particular kinase mutation may help to define the initial clinical course of a GIST. Based on retrospective studies from time periods that predated the clinical use of kinase inhibitors, current recommendations for assessing the risk of progression for a newly diagnosed primary GIST rely on three parameters (see Table 1):[2,23,24,25,26]
- Mitotic index (mitoses per 50 high-power fields).
- Tumor size.
- Tumor location.
Table 1. Risk Stratification of Primary GIST by Mitotic Index, Tumor Size, and Tumor Locationa
| GIST = gastrointestinal stromal tumors; hpf = high-power field, assessed from an area that on initial screen appears to have the highest mitotic activity. |
| a Annual review of pathology by ANNUAL REVIEWS, INC. Reproduced with permission of ANNUAL REVIEWS, INC., in the format Internet posting via Copyright Clearance Center.[2] |
| b Small numbers of cases. |
| Mitotic Index, hpf |
Size, cm |
Site and Risk of Progressive Disease (%) |
| Gastric |
Jejunum/Ileum |
Duodenum |
Rectum |
| ≤5 per 50 |
≤2 |
None (0) |
None (0) |
None (0) |
None (0) |
| >2 ≤5 |
Very low (1.9) |
Low (4.3) |
Low (8.3) |
Low (8.5) |
| >5 ≤10 |
Low (3.6) |
Moderate (24) |
(Insufficient data) |
(Insufficient data) |
| >10 |
Moderate (12) |
High (52) |
High (34) |
High (57)b |
| >5 per 50 |
≤2 |
Noneb |
Highb |
(Insufficient data) |
High (54) |
| >2 ≤5 |
Moderate (16) |
High (73) |
High (50) |
High (52) |
| >5 ≤10 |
High (55) |
High (85) |
(Insufficient data) |
(Insufficient data) |
| >10 |
High (86) |
High (90) |
High (86) |
High (71) |
Survival
Compared to other intra-abdominal sarcomas, survival in GIST patients after surgery alone is favorable.[27] In a retrospective study involving 200 patients that predated the use of TKI, the 5-year disease-specific survival rate for GIST patients with primary disease who underwent complete resection of gross disease (N = 80) was 54%, with survival predicted by tumor size; the overall disease-specific survival was 35% at 5 years.[11] Other studies, which also predated TKI, reported 5-year survival rates of 40% to 63% for patients undergoing complete resections of GIST.
In the retrospective study of 200 patients cited in Table 1 above, 7% had isolated local recurrence and 47% had metastasis.[11] The site of relapse for GIST is usually intra-abdominal, involving the peritoneum, the liver, or both; true local recurrences are uncommon, and typically there is widespread intraperitoneal recurrence that may not be detectable by imaging techniques.[27] The median disease-specific survival of patients with metastatic GIST (N = 94) was 19 months.[11] In one retrospective study involving 119 patients with metastatic GIST, it was found that once a GIST becomes metastatic, kinase genotype did not factor into overall survival.[28]
The median time to recurrence for patients on imatinib is 2 years.[27]
Follow-up
The most appropriate tests and frequency of testing for metastatic or recurrent disease in patients who have undergone GIST resection are ill-defined, since the impact of follow-up strategies on clinical outcomes is not known. Follow-up recommendations are, therefore, based upon expert opinion and clinical judgment taking into account tumor site, size, and mitotic index. For surgically treated patients with localized disease, routine follow-up schedules may differ across institutions and may depend on the risk status of the tumor.[18] Abdominal/pelvic CT may be performed every 3 to 6 months, but very low-risk lesions may not need routine follow-up testing.[18]
CT or 18F-FDG PET are used to monitor therapeutic effects in patients receiving systemic therapy for unresectable, metastatic, or recurrent disease.[27] 18F-FDG PET may also be helpful in detecting resistance to TKI. If 18F-FDG PET is used to monitor therapy with a TKI, a baseline FDG PET is often performed before kinase inhibitor administration. Because 18F-FDG PET imaging may detect the activity of imatinib in GIST much earlier than CT imaging, imaging of GIST with 18F-FDG PET may represent a useful diagnostic modality for the very early assessment of response to imatinib therapy; a decrease in tumor avidity for 18F-FDG may be detected as early as 24 hours after a single dose of imatinib.[12]
Related Summary
(Refer to the Abdominal Cancers section in the PDQ summary on Unusual Cancers of Childhood Treatment for information on gastrointestinal stromal tumors in children.)
References:
- Judson I, Demetri G: Advances in the treatment of gastrointestinal stromal tumours. Ann Oncol 18 (Suppl 10): x20-4, 2007.
- Corless CL, Heinrich MC: Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol 3: 557-86, 2008.
- Tran T, Davila JA, El-Serag HB: The epidemiology of malignant gastrointestinal stromal tumors: an analysis of 1,458 cases from 1992 to 2000. Am J Gastroenterol 100 (1): 162-8, 2005.
- Kawanowa K, Sakuma Y, Sakurai S, et al.: High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol 37 (12): 1527-35, 2006.
- Agaimy A, Wünsch PH, Hofstaedter F, et al.: Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol 31 (1): 113-20, 2007.
- Nowain A, Bhakta H, Pais S, et al.: Gastrointestinal stromal tumors: clinical profile, pathogenesis, treatment strategies and prognosis. J Gastroenterol Hepatol 20 (6): 818-24, 2005.
- Gastrointestinal stromal tumor. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 175-80.
- Corless CL, Fletcher JA, Heinrich MC: Biology of gastrointestinal stromal tumors. J Clin Oncol 22 (18): 3813-25, 2004.
- Joensuu H: Gastrointestinal stromal tumor (GIST). Ann Oncol 17 (Suppl 10): x280-6, 2006.
- Miettinen M, Lasota J: Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med 130 (10): 1466-78, 2006.
- DeMatteo RP, Lewis JJ, Leung D, et al.: Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg 231 (1): 51-8, 2000.
- Demetri GD: Gastrointestinal stromal tumor. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1060-73.
- Nickl NJ: Gastrointestinal stromal tumors: new progress, new questions. Curr Opin Gastroenterol 20 (5): 482-7, 2004.
- Vander Noot MR 3rd, Eloubeidi MA, Chen VK, et al.: Diagnosis of gastrointestinal tract lesions by endoscopic ultrasound-guided fine-needle aspiration biopsy. Cancer 102 (3): 157-63, 2004.
- Hirota S, Isozaki K, Moriyama Y, et al.: Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 279 (5350): 577-80, 1998.
- Kindblom LG, Remotti HE, Aldenborg F, et al.: Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol 152 (5): 1259-69, 1998.
- Antonescu CR: Targeted therapy of cancer: new roles for pathologists in identifying GISTs and other sarcomas. Mod Pathol 21 (Suppl 2): S31-6, 2008.
- Casali PG, Jost L, Reichardt P, et al.: Gastrointestinal stromal tumors: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol 19 (Suppl 2): ii35-8, 2008.
- Heinrich MC, Corless CL, Duensing A, et al.: PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299 (5607): 708-10, 2003.
- Andersson J, Sihto H, Meis-Kindblom JM, et al.: NF1-associated gastrointestinal stromal tumors have unique clinical, phenotypic, and genotypic characteristics. Am J Surg Pathol 29 (9): 1170-6, 2005.
- Agaimy A, Pelz AF, Corless CL, et al.: Epithelioid gastric stromal tumours of the antrum in young females with the Carney triad: a report of three new cases with mutational analysis and comparative genomic hybridization. Oncol Rep 18 (1): 9-15, 2007.
- Carney JA: Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc 74 (6): 543-52, 1999.
- Miettinen M, Sobin LH, Lasota J: Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol 29 (1): 52-68, 2005.
- Miettinen M, Makhlouf H, Sobin LH, et al.: Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol 30 (4): 477-89, 2006.
- Miettinen M, Kopczynski J, Makhlouf HR, et al.: Gastrointestinal stromal tumors, intramural leiomyomas, and leiomyosarcomas in the duodenum: a clinicopathologic, immunohistochemical, and molecular genetic study of 167 cases. Am J Surg Pathol 27 (5): 625-41, 2003.
- Miettinen M, Furlong M, Sarlomo-Rikala M, et al.: Gastrointestinal stromal tumors, intramural leiomyomas, and leiomyosarcomas in the rectum and anus: a clinicopathologic, immunohistochemical, and molecular genetic study of 144 cases. Am J Surg Pathol 25 (9): 1121-33, 2001.
- Demetri GD, Benjamin RS, Blanke CD, et al.: NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST)–update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw 5 (Suppl 2): S1-29; quiz S30, 2007.
- Gold JS, van der Zwan SM, Gönen M, et al.: Outcome of metastatic GIST in the era before tyrosine kinase inhibitors. Ann Surg Oncol 14 (1): 134-42, 2007.
Cellular and Molecular Classifications of GIST
Gastrointestinal stromal tumors (GIST) appear to originate from interstitial cells of Cajal (ICC) or their stem cell-like precursors.[1,2] ICC are pacemaker-like intermediates between the GI autonomic nervous system and smooth muscle cells regulating GI motility and autonomic nerve function.[3,4] KIT-positive and KIT-dependent, ICC are located around the myenteric plexus and the muscularis propria throughout the GI tract. ICC or their stem cell-like precursors can differentiate into smooth muscle cells if KIT signaling is disrupted.[5]
Approximately 95% of GIST are positive for the CD117 antigen, an epitope of KIT receptor tyrosine kinase expressed by ICC.[6] However, CD117 immunohistochemistry is not specific for GIST, as weak reactivity occurs with other mesenchymal neoplasms. Accordingly, CD117 immunostaining of tumors should be interpreted cautiously in the context of other immunomarkers and the anatomic location and morphology of the tumor in order to differentiate GIST from other mesenchymal, neural, and neuroendocrine neoplasms.[6] Immunohistochemical staining for protein kinase C theta and DOG1 may help distinguish GIST from other mesenchymal tumors, particularly those that are KIT-negative.[6,7,8,9] DOG1 (discovered on GIST 1) is a protein of unknown function that is expressed strongly on GIST and is rarely expressed on other soft tissue tumors.[9]
Approximately 85% of GIST contain oncogenic mutations in one of two receptor tyrosine kinases: KIT or platelet-derived growth factor receptor alpha (PDGFRA).[6] Constitutive activation of either of these receptor tyrosine kinases plays a central role in the pathogenesis of GIST.[1,10] The proper identification of GIST with genotyping is very important because of the availability of specific, molecular-targeted therapy with KIT/PDGFRA tyrosine kinase inhibitors (TKI), such as imatinib mesylate or, in the case of imatinib-resistant GIST, sunitinib malate.[11,12,13]
GIST may fall into one or more of the following subgroups:
- KIT-mutant GIST. Approximately 80% of all GIST contain a mutation in the KIT receptor tyrosine kinase that results in constitutive activation of the protein.[6] The KIT gene maps to 4q12-13, in the vicinity of genes encoding the receptor tyrosine kinases PDGFRA and vascular endothelial growth factor receptor 2 (VEGFR2).[14] Mutations in five different KIT exons have been observed in GIST: exon 11 (67%), exon 9 (10%), and exons 8, 13, and 17 (3%).[6,12] Typically, GIST are heterozygous for a particular mutation, but loss of the remaining wild-type KIT allele occurs in approximately 8% to 15% of tumors and may be associated with malignant progression.[12,15,16]KIT mutation variants exhibit distinct anatomic distributions: exon 8 (small bowel), exon 9 (small bowel, colon), and exons 11, 13, and 17 (all sites).[6]KIT-mutant tumors express protein kinase C theta and DOG1, a distinguishing feature of mesenchymal tumors.[8,9,17]
- PDGFRA-mutant GIST. Approximately 5% to 8% of GIST harbor a mutation in PDGFRA, a close homolog of KIT with similar extracellular and cytoplasmic domains.[10]PDGFRA-mutant GIST may differ from KIT-mutant GIST in a number of ways, including a marked predilection for the stomach, epithelioid morphology, myxoid stroma, nuclear pleomorphism, and variable expression of CD117.[17,18,19,20,21,22] As with KIT-mutant GIST, PDGFRA-mutant tumors express protein kinase C theta and DOG1.[8,9,18]
- Wild-type GIST. Wild-type GIST comprise approximately 12% to 15% of all GIST. In these tumors no detectable mutations have been identified in either KIT or PDGFRA, although KIT is still phosphorylated. For these tumors, there is no particular association with anatomic location or clinical outcome.[6]
- KIT-negative GIST. In approximately 5% of GIST, immunohistochemistry (IHC) for CD117 is completely negative or uncertain; in these instances, IHC may lack sufficient sensitivity to detect small amounts of mutant kinase.[6] Approximately 30% of these tumors harbor PDGFRA gene mutations while more than half have KIT mutations.[6,18,19,22]
- GIST syndromes. In adults, syndromic GIST have been associated with neurofibromatosis type 1 (NF1) and the Carney triad (gastric epithelioid GIST, extra-adrenal paraganglioma, and pulmonary chondroma).[23,24,25]
- NF1-associated GIST have a propensity for multicentricity within the GI tract and spindle cell morphology and do not harbor KIT or PDGFRA gene mutations. NF1-associated GIST are typically positive for the CD117 antigen.[24]
- Carney triad syndrome-associated GIST are predominantly of epithelioid morphology, tend to occur in the antrum, lack conventional KIT and PDGRFA gene mutations, and tend to run an indolent course.[6,23,25]
- Familial GIST. As of 2008, approximately two dozen kindreds with heritable mutations in KIT or PFGFRA have been identified. Penetrance in these kindreds is high, with most affected members developing one or more GIST by middle age; however, in many patients the tumors follow a benign course.[6]
- Multiple GIST. Although rare, multiple GIST have been observed in patients with NF1 and germline KIT gene mutations.[26] In addition, sporadic, synchronous, and metachronous tumors have been observed in patients without identifiable germline risk factors, suggesting that other genes that predispose to the development of GIST have yet to be discovered.[6,26]
- Secondary GIST mutations acquired during imatinib therapy. Metastatic disease with acquired drug resistance, usually the result of secondary, imatinib-resistant mutations in KIT or PDGFRA tyrosine kinase domains I and II, can occur during imatinib treatment.[16,27,28,29]
References:
- Hirota S, Isozaki K, Moriyama Y, et al.: Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 279 (5350): 577-80, 1998.
- Kindblom LG, Remotti HE, Aldenborg F, et al.: Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol 152 (5): 1259-69, 1998.
- Maeda H, Yamagata A, Nishikawa S, et al.: Requirement of c-kit for development of intestinal pacemaker system. Development 116 (2): 369-75, 1992.
- Huizinga JD, Thuneberg L, Klüppel M, et al.: W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature 373 (6512): 347-9, 1995.
- Torihashi S, Nishi K, Tokutomi Y, et al.: Blockade of kit signaling induces transdifferentiation of interstitial cells of cajal to a smooth muscle phenotype. Gastroenterology 117 (1): 140-8, 1999.
- Corless CL, Heinrich MC: Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol 3: 557-86, 2008.
- Blay P, Astudillo A, Buesa JM, et al.: Protein kinase C theta is highly expressed in gastrointestinal stromal tumors but not in other mesenchymal neoplasias. Clin Cancer Res 10 (12 Pt 1): 4089-95, 2004.
- Duensing A, Joseph NE, Medeiros F, et al.: Protein Kinase C theta (PKCtheta) expression and constitutive activation in gastrointestinal stromal tumors (GISTs). Cancer Res 64 (15): 5127-31, 2004.
- West RB, Corless CL, Chen X, et al.: The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol 165 (1): 107-13, 2004.
- Heinrich MC, Corless CL, Duensing A, et al.: PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299 (5607): 708-10, 2003.
- Judson I, Demetri G: Advances in the treatment of gastrointestinal stromal tumours. Ann Oncol 18 (Suppl 10): x20-4, 2007.
- Heinrich MC, Corless CL, Demetri GD, et al.: Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 21 (23): 4342-9, 2003.
- Demetri GD, von Mehren M, Blanke CD, et al.: Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 347 (7): 472-80, 2002.
- Stenman G, Eriksson A, Claesson-Welsh L: Human PDGFA receptor gene maps to the same region on chromosome 4 as the KIT oncogene. Genes Chromosomes Cancer 1 (2): 155-8, 1989.
- O’Riain C, Corless CL, Heinrich MC, et al.: Gastrointestinal stromal tumors: insights from a new familial GIST kindred with unusual genetic and pathologic features. Am J Surg Pathol 29 (12): 1680-3, 2005.
- Antonescu CR, Besmer P, Guo T, et al.: Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 11 (11): 4182-90, 2005.
- Wasag B, Debiec-Rychter M, Pauwels P, et al.: Differential expression of KIT/PDGFRA mutant isoforms in epithelioid and mixed variants of gastrointestinal stromal tumors depends predominantly on the tumor site. Mod Pathol 17 (8): 889-94, 2004.
- Debiec-Rychter M, Wasag B, Stul M, et al.: Gastrointestinal stromal tumours (GISTs) negative for KIT (CD117 antigen) immunoreactivity. J Pathol 202 (4): 430-8, 2004.
- Medeiros F, Corless CL, Duensing A, et al.: KIT-negative gastrointestinal stromal tumors: proof of concept and therapeutic implications. Am J Surg Pathol 28 (7): 889-94, 2004.
- Sakurai S, Hasegawa T, Sakuma Y, et al.: Myxoid epithelioid gastrointestinal stromal tumor (GIST) with mast cell infiltrations: a subtype of GIST with mutations of platelet-derived growth factor receptor alpha gene. Hum Pathol 35 (10): 1223-30, 2004.
- Wardelmann E, Hrychyk A, Merkelbach-Bruse S, et al.: Association of platelet-derived growth factor receptor alpha mutations with gastric primary site and epithelioid or mixed cell morphology in gastrointestinal stromal tumors. J Mol Diagn 6 (3): 197-204, 2004.
- Pauls K, Merkelbach-Bruse S, Thal D, et al.: PDGFRalpha- and c-kit-mutated gastrointestinal stromal tumours (GISTs) are characterized by distinctive histological and immunohistochemical features. Histopathology 46 (2): 166-75, 2005.
- Carney JA: Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc 74 (6): 543-52, 1999.
- Andersson J, Sihto H, Meis-Kindblom JM, et al.: NF1-associated gastrointestinal stromal tumors have unique clinical, phenotypic, and genotypic characteristics. Am J Surg Pathol 29 (9): 1170-6, 2005.
- Agaimy A, Pelz AF, Corless CL, et al.: Epithelioid gastric stromal tumours of the antrum in young females with the Carney triad: a report of three new cases with mutational analysis and comparative genomic hybridization. Oncol Rep 18 (1): 9-15, 2007.
- Kang DY, Park CK, Choi JS, et al.: Multiple gastrointestinal stromal tumors: Clinicopathologic and genetic analysis of 12 patients. Am J Surg Pathol 31 (2): 224-32, 2007.
- Tamborini E, Bonadiman L, Greco A, et al.: A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology 127 (1): 294-9, 2004.
- Chen LL, Sabripour M, Andtbacka RH, et al.: Imatinib resistance in gastrointestinal stromal tumors. Curr Oncol Rep 7 (4): 293-9, 2005.
- Debiec-Rychter M, Cools J, Dumez H, et al.: Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology 128 (2): 270-9, 2005.
Stage Information for GIST
Note: The American Joint Committee on Cancer (AJCC) has published the 8th edition of the AJCC Cancer Staging Manual, which includes revisions to the staging for this disease. Implementation of the 8th edition began in January 2018. The PDQ Adult Treatment Editorial Board, which maintains this summary, is reviewing the revised staging and will make appropriate changes as needed.
A formal staging system for gastrointestinal stromal tumors (GIST) appeared for the first time in the 7th edition of the Staging Manual.[1] Because of the limited availability of mutation testing, mutation status is not included in the AJCC system. In addition, since nodal involvement is so rare, and nodal dissection is not indicated, the designation N0/pN0 should be used in the absence of information about the regional nodes. Note that treatment decisions may not be based strictly on the stage of disease.[2,3,4]
AJCC Stage Groupings and TNM Definitions (for GIST at All Sites)
This staging system for GIST is new for the 7th edition of the AJCC Cancer Staging Manual.[1]
Table 2. Primary Tumor (T)a
| a Reprinted with permission from AJCC: Gastrointestinal stromal tumor. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 181-9. |
| TX |
Primary tumor cannot be assessed. |
| T0 |
No evidence for primary tumor. |
| T1 |
Tumor ≤2 cm. |
| T2 |
Tumor >2 cm but not >5 cm. |
| T3 |
Tumor >5 cm but not >10 cm. |
| T4 |
Tumor >10 cm in greatest dimension. |
Table 3. Regional Lymph Nodes (N)a
| a Reprinted with permission from AJCC: Gastrointestinal stromal tumor. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 181-9. |
| NX |
Regional lymph nodes cannot be assessed. |
| N0 |
No regional lymph node metastasis. |
| N1 |
Regional lymph node metastasis. |
Table 4. Distant Metastasis (M)a
| a Reprinted with permission from AJCC: Gastrointestinal stromal tumor. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 181-9. |
| M0 |
No distant metastasis. |
| M1 |
Distant metastasis. |
Table 5. Anatomic Stage/Prognostic Groupsa
| gist = gastrointestinal stromal tumors. |
| a Reprinted with permission from AJCC: Gastrointestinal stromal tumor. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 181-9. |
| b Also to be used for omentum. |
| c Also to be used for esophagus, colorectal, mesentery, and peritoneum. |
| Gastric GISTb |
| Group |
T |
N |
M |
Mitotic Rate |
| Stage IA |
T1 or T2 |
N0 |
M0 |
Low |
| Stage IB |
T3 |
N0 |
M0 |
Low |
| Stage II |
T1 |
N0 |
M0 |
High |
| T2 |
N0 |
M0 |
High |
| T4 |
N0 |
M0 |
Low |
| Stage IIIA |
T3 |
N0 |
M0 |
High |
| Stage IIIB |
T4 |
N0 |
M0 |
High |
| Stage IV |
Any T |
N1 |
M0 |
Any rate |
| Any T |
Any N |
M1 |
Any rate |
| Small Intestinal GISTc |
| Group |
T |
N |
M |
Mitotic rate |
| Stage I |
T1 or T2 |
N0 |
M0 |
Low |
| Stage II |
T3 |
N0 |
M0 |
Low |
| Stage IIIA |
T1 |
N0 |
M0 |
High |
| T4 |
N0 |
M0 |
Low |
| Stage IIIB |
T2 |
N0 |
M0 |
High |
| T3 |
N0 |
M0 |
High |
| T4 |
N0 |
M0 |
High |
| Stage IV |
Any T |
N1 |
M0 |
Any rate |
| Any T |
Any N |
M1 |
Any rate |
References:
- Gastrointestinal stromal tumor. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 175-80.
- Demetri GD: Gastrointestinal stromal tumor. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1060-73.
- Demetri GD, Benjamin RS, Blanke CD, et al.: NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST)–update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw 5 (Suppl 2): S1-29; quiz S30, 2007.
- Casali PG, Jost L, Reichardt P, et al.: Gastrointestinal stromal tumors: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol 19 (Suppl 2): ii35-8, 2008.
Treatment Option Overview for GIST
The management of patients with gastrointestinal stromal tumors (GIST) is a multidisciplinary effort involving close collaboration between pathologists, medical oncologists, surgeons, and imaging experts.[1]
Treatment may involve surgery and/or the use of tyrosine kinase inhibitors (TKI) depending on the extent of disease and tumor sensitivity to TKI. Although recurrence is common for patients with high-risk tumors (see Table1), complete resection of localized tumors may be associated with long-term disease-free survival (DFS).[2][Level of evidence: 3iiiDii] Standard chemotherapy is not used because of the insensitivity of GIST to chemotherapeutic agents.[3,4,5,6] Radiation therapy rarely has a role in the management of patients with GIST; it may occasionally be used for pain control in patients with limited, bulky hepatic metastases or with a single, large metastatic lesion fixed to the abdominal or pelvic wall.[1][Level of evidence: 2Div] Whether tumors 2 cm or smaller with a mitotic index of 5 or less per 50 high-power fields require surgery is controversial. Such tumors appear to have low rates of progression and metastasis,[7] but the absolute rates are not known with precision. Endoscopic surveillance is an option.
Surgical Therapy
Surgery is typically the initial therapy for the following types of patients:
- Those with primary GIST who do not have evidence of metastasis.
- Those with tumors that are technically resectable if the risks of morbidity are acceptable.
In the surgical treatment of GIST, the goal is complete gross resection with an intact pseudocapsule and negative microscopic margins.[4] Because lymph node metastasis is rare with GIST, lymphadenectomy of clinically uninvolved nodes is not necessary.
Although a prospective, randomized trial studying the role of laparoscopic surgery in the management of GIST has not been performed, several studies, listed below, indicate a role for this surgical approach with gastric tumors:
- In one retrospective study involving 33 patients with gastric tumors ranging in size from 0.5 cm to 10.5.cm, all gross tumors could be successfully removed by laparoscopic surgery, with short hospitalizations and low morbidity. There were no recurrences observed with a mean follow-up of 13 months.[8][Level of evidence: 3iiDii]
- In another retrospective study involving 60 patients, laparoscopic or laparoscopy-assisted resections of GIST measuring 2 cm to 5 cm were associated with a 5-year DFS of 100% for very low-risk groups and low-risk groups versus a 5-year DFS of 89.9% for intermediate-risk groups and high-risk groups; no local or distant recurrences were observed for tumors smaller than 4 cm in size.[9][Level of evidence: 3iiDi]
- In another study involving 50 consecutive patients undergoing laparoscopic or laparoendoscopic resection of gastric GIST (mean tumor size = 4.4 cm) who were identified in a prospectively collected database, 46 (92%) patients were found to be disease free at a mean follow-up of 36 months.[10][Level of evidence: 3iiDi]
Neoadjuvant imatinib therapy can be used for patients with large tumors or difficult-to-access small GIST that are considered marginally resectable. In addition, patients with primary localized GIST deemed unresectable are often treated with imatinib.[4,11]
Chemotherapy
Before the advent of molecularly targeted therapy with TKI, efforts to treat GIST with conventional cytotoxic chemotherapy were essentially futile.[1] The extreme resistance of GIST to chemotherapy may be caused, in part, by the increased expression of P-glycoprotein, the product of the MDR-1 (multidrug resistance-1) gene, and MRP1 (multidrug resistance protein-1), which are cellular efflux pumps that may prevent chemotherapeutic agents from reaching therapeutic intracellular concentrations in GIST cells.[1,12] There is universal agreement that standard chemotherapy has no role in the primary therapy of GIST.[4,5,6]
Tyrosine Kinase Inhibitor Therapy
TKIs have revolutionized the management of GIST. The TKI imatinib mesylate is used as the first-line treatment for unresectable, metastatic, or recurrent GIST. Although complete responses are rare, a large majority of patients with metastatic or inoperable GIST have either a partial response or disease stabilization after starting imatinib. Median survival rates have gone from less than 2 years to more than 5 years since the advent of imatinib therapy.[13]
Therapy with neoadjuvant imatinib to reduce the tumor volume may be used for patients with very large primary GIST that cannot be removed without the risk of unacceptable morbidity.[11] Additional therapy with adjuvant imatinib is being studied to determine whether imatinib reduces recurrence, which is common after resection of primary GIST.[4]
Because disease progression has been reported to follow the cessation of imatinib therapy, patients with unresectable or metastatic disease are often treated with a TKI indefinitely, as long as the disease does not progress and patient tolerance permits.[1,14] In a multicenter trial in which 58 patients with advanced GIST who had disease stability after at least 1 year of imatinib therapy were randomly assigned to continue (n = 26) or to discontinue (n = 32) imatinib (with reinstitution for progression), 8 and 26 patients progressed at a median of 18 and 6.1 months, respectively (P < .0001). However, 24 of the 26 patients in the latter group responded again to reinstitution of imatinib.[14][Level of evidence: 1iiDiii] There were no differences in overall survival (OS), development of imatinib resistance, or quality of life between the two groups.[14][Level of evidence: 1iiA and 1iiC]
Drug dose and schedule
A patient with unresectable or metastatic GIST may be treated with an initial dose of 400 mg imatinib mesylate daily, with therapeutic effects monitored by fluorine F 18-fludeoxyglucose positron emission tomography (18F-FDG PET) or computed tomography; dose escalation to 400 mg twice a day may be appropriate for patients with progressive disease, although it is unlikely to help patients who progress within 2 months of initiation of imatinib therapy.[4,15,16,17] An initial dose of 800 mg daily may be appropriate for patients with GIST harboring KIT exon 9 mutations.[18] Resistance to imatinib may be primary with rapid progression of disease despite an increase in the imatinib dose, although this appears to occur in fewer than 20% of patients; some investigators have speculated that GIST with primary resistance to imatinib have mutations in other oncogenic signaling pathways that do not involve KIT.[1,19,20]
The majority of patients treated with imatinib ultimately experience disease progression after an initial response because of the development of delayed imatinib resistance. In most cases, delayed resistance is associated with secondary mutations in a separate portion of the KIT-coding sequence.[20,21]
The oral TKI sunitinib malate is generally given to patients with unresectable disease who progress on higher-dose imatinib, although individuals with localized progression may be candidates for resection.[22] Less specific than imatinib, sunitinib inhibits vascular endothelial growth factor receptors (VEGFR 1-3), Fms-like tyrosine kinase-3 (FLT3), colony-stimulating factor 1 receptor (CSF-1R), and RET as well as KIT and PDGFR and displays antiangiogenic activity.[23,24,25] A number of other targeted therapeutics for the treatment of GIST are in development, including a variety of other kinase inhibitors, heat-shock protein 90 (Hsp90) inhibitors such as IPI-504, the mTOR inhibitor RAD001, and histone deacetylase inhibitors.[3,26]
Treatment with imatinib or sunitinib may be continued for as long as the patient appears to be deriving clinical benefit or has disease stability.[4]
Response to kinase inhibitors
KIT- and PDGFRA-mutational analysis may be of help in predicting responses to kinase inhibitors for patients with unresectable, metastatic, or recurrent GIST who are undergoing therapy with selective TKIs.[18,27,28,29,30] However, the data are preliminary and mutational analysis for treatment decisions is not routine. There is currently no evidence that basing treatment decisions on mutational analysis improves OS. Four trials involving 768 patients and imatinib doses ranging from 400 mg to 800 mg per day have correlated tumor genotypes and complete and partial objective responses (see Table 6).[3] For these 768 genotyped GIST, the objective response for KIT exon 11 mutant, KIT exon 9 mutant, and wild-type (no KIT or PDGFRA mutation) GIST were 71%, 38%, and 28% (weighted averages), respectively; rates of primary resistance to imatinib therapy were 5%, 16%, and 23%, respectively.
Table 6. Relationship Between Tyrosine Kinase Genotype and Response to Imatinib Therapya
| |
European Phase I/IIb Trial, % (n)[31] |
B-2222 Phase IIc Trial, % (n)[32] |
European/Australasian Phase IIId Trial, % (n)[18] |
North American Phase IIIe Trial, % (n)[33] |
| N = number in sample, number of observations; NR = not reported. |
| a Annual review of pathology by ANNUAL REVIEWS, INC. Reproduced with permission of ANNUAL REVIEWS, INC., in the format Internet posting via Copyright Clearance Center.[3] |
| b[Level of evidence: 1iiA, 1iiDii,1iiDiv (Phase 1) and2A; 2Div(Phase II)] |
| c[Level of evidence: 1iiDiv] |
| d[Level of evidence: 1iiAand1iiDiii] |
| e[Level of evidence: 1iiDiv] |
| f Defined as complete or partial response by Southwest Oncology Group (SWOG) criteria for B-2222 or RECIST (Response EvaluationCriteria in Solid Tumors) for all other trials; excludes nonevaluable patients. |
| g Statistically significant difference compared withKIT9 and wild-type (noKITorPDGFRAmutation) groups. |
| Study participants |
(N = 37) |
(N = 127) |
(N= 377) |
(N = 324) |
| Objective responsef |
|
| KITexon 11 |
83 (24) |
83g(85) |
70g(248) |
67g(211) |
| KIT exon 9 |
25 (4) |
48 (23) |
35 (58) |
40 (25) |
| Wild-type |
33 (6) |
0 (9) |
25 (52) |
39 (33) |
| Progressive disease |
|
| KIT exon 11 |
4 |
5 |
3 |
NR |
| KIT exon 9 |
0 |
17 |
17 |
NR |
| Wild-type |
33 |
56 |
19 |
NR |
Kinase genotype appears to correlate with progression-free survival (PFS) and OS. The median time to tumor progression (TTP) for patients whose GIST harbors a KIT exon 11 mutation has been reported to be more than 1 year longer than the median TTP for patients whose tumors have KIT exon 9 or wild-type kinase genotypes; a similar OS benefit has been reported for patients with KIT exon 11 mutations versus the other common genotype subsets.[3] In a subset analysis of the European/Australasian phase III trial, it was found that the PFS of GIST patients with KIT exon 9 mutations was significantly better when patients were treated with 800 mg of imatinib per day as compared with 400 mg per day (P = .0013), with a reduction of relative risk of 61%.[18][Level of evidence: 1iiDiii] Accordingly, routine tumor typing and imatinib dose selection based on the presence or absence of a KIT exon 9 mutation is recommended by some but not all GIST experts.[3,4]
Drug side effects and other considerations
The most common toxicities associated with imatinib therapy, all of which may improve with prolonged treatment, include the following:[5,16,17,34,35]
- Fluid retention (especially periorbital edema or peripheral edema; occasionally pleural effusion or ascites).
- Diarrhea.
- Nausea (may be diminished if taken with food).
- Fatigue.
- Muscle cramps.
- Abdominal pain.
- Rash.
- Mild (macrocytic) anemia.
(Refer to the PDQ summaries on Lymphedema [edema], Gastrointestinal Complications [diarrhea], Treatment-Related Nausea and Vomiting, Fatigue [fatigue and anemia], and Cancer Pain for more information on some of the conditions listed above.)
Treatment with sunitinib may be considered for patients with life-threatening side effects from imatinib that cannot be managed by maximum supportive care.[4] Common side effects associated with sunitinib therapy include the following:[22,36]
- Fatigue.
- Nausea and vomiting.
- Anemia.
- Neutropenia.
- Diarrhea.
- Abdominal pain.
- Mucositis.
- Anorexia.
- Skin or hair discoloration.
- Hypothyroidism (thyroid function monitoring with TSH is generally recommended to detect subclinical hypothyroidism).
(Refer to the PDQ summary on Nutrition in Cancer Care for more information on some of the conditions listed above.)
Less frequent toxicities include bleeding, fever, and hand-foot syndrome.[22]
Therapy with sunitinib also may be cardiotoxic. In a retrospective study of a phase I/II trial studying the efficacy of sunitinib in treating imatinib-resistant, metastatic GIST, 8% of 75 patients who received repeating cycles of sunitinib experienced congestive heart failure while 47% developed hypertension (>150 per 100 mm Hg); reductions in left ventricular ejection fraction were at least 10% in 28% of patients.[37][Level of evidence: 3iiB]
A number of other drugs and certain fruit juices (e.g., grapefruit, pomegranate) may alter plasma levels of imatinib or sunitinib by inducing or inhibiting cytochrome P450 isoenzyme 3A4 (CYP450 3A4), the primary enzyme involved in the metabolism of these TKIs.[4,38,39,40,41,42] For patients taking drugs that affect CYP450 3A4 levels, dose modification of the TKI or substitution with medications that do not affect CYP450 3A4 may be necessary.
References:
- Demetri GD: Gastrointestinal stromal tumor. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1060-73.
- Judson I, Demetri G: Advances in the treatment of gastrointestinal stromal tumours. Ann Oncol 18 (Suppl 10): x20-4, 2007.
- Corless CL, Heinrich MC: Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol 3: 557-86, 2008.
- Demetri GD, Benjamin RS, Blanke CD, et al.: NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST)–update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw 5 (Suppl 2): S1-29; quiz S30, 2007.
- Demetri GD, von Mehren M, Blanke CD, et al.: Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 347 (7): 472-80, 2002.
- Edmonson JH, Marks RS, Buckner JC, et al.: Contrast of response to dacarbazine, mitomycin, doxorubicin, and cisplatin (DMAP) plus GM-CSF between patients with advanced malignant gastrointestinal stromal tumors and patients with other advanced leiomyosarcomas. Cancer Invest 20 (5-6): 605-12, 2002.
- Miettinen M, Sobin LH, Lasota J: Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol 29 (1): 52-68, 2005.
- Huguet KL, Rush RM Jr, Tessier DJ, et al.: Laparoscopic gastric gastrointestinal stromal tumor resection: the mayo clinic experience. Arch Surg 143 (6): 587-90; discussion 591, 2008.
- Otani Y, Furukawa T, Yoshida M, et al.: Operative indications for relatively small (2-5 cm) gastrointestinal stromal tumor of the stomach based on analysis of 60 operated cases. Surgery 139 (4): 484-92, 2006.
- Novitsky YW, Kercher KW, Sing RF, et al.: Long-term outcomes of laparoscopic resection of gastric gastrointestinal stromal tumors. Ann Surg 243 (6): 738-45; discussion 745-7, 2006.
- Bonvalot S, Eldweny H, Péchoux CL, et al.: Impact of surgery on advanced gastrointestinal stromal tumors (GIST) in the imatinib era. Ann Surg Oncol 13 (12): 1596-603, 2006.
- Plaat BE, Hollema H, Molenaar WM, et al.: Soft tissue leiomyosarcomas and malignant gastrointestinal stromal tumors: differences in clinical outcome and expression of multidrug resistance proteins. J Clin Oncol 18 (18): 3211-20, 2000.
- Blanke CD, Demetri GD, von Mehren M, et al.: Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol 26 (4): 620-5, 2008.
- Blay JY, Le Cesne A, Ray-Coquard I, et al.: Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: the French Sarcoma Group. J Clin Oncol 25 (9): 1107-13, 2007.
- Verweij J, Casali PG, Zalcberg J, et al.: Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet 364 (9440): 1127-34, 2004.
- Verweij J, van Oosterom A, Blay JY, et al.: Imatinib mesylate (STI-571 Glivec, Gleevec) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Results from an EORTC Soft Tissue and Bone Sarcoma Group phase II study. Eur J Cancer 39 (14): 2006-11, 2003.
- Benjamin RS, Rankin C, Fletcher C, et al.: Phase III dose-randomized study of imatinib mesylate (STI571) for GIST: Intergroup S0033 early results. [Abstract] Proceedings of the American Society of Clinical Oncology 22: A-3271, 2003.
- Debiec-Rychter M, Sciot R, Le Cesne A, et al.: KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 42 (8): 1093-103, 2006.
- Kindblom LG, Meis-Kindblom J, Bümming P, et al.: Incidence, prevalence, phenotype and biologic spectrum of gastrointestinal stromal cell tumors (GIST): a population-based study of 600 cases. [Abstract] Ann Oncol 13 (Suppl 5): A-577O, 157, 2002..
- Gramza AW, Corless CL, Heinrich MC: Resistance to Tyrosine Kinase Inhibitors in Gastrointestinal Stromal Tumors. Clin Cancer Res 15 (24): 7510-7518, 2009.
- Desai J, Shankar S, Heinrich MC, et al.: Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res 13 (18 Pt 1): 5398-405, 2007.
- Demetri GD, van Oosterom AT, Garrett CR, et al.: Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 368 (9544): 1329-38, 2006.
- O’Farrell AM, Abrams TJ, Yuen HA, et al.: SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 101 (9): 3597-605, 2003.
- Mendel DB, Laird AD, Xin X, et al.: In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 9 (1): 327-37, 2003.
- Murray LJ, Abrams TJ, Long KR, et al.: SU11248 inhibits tumor growth and CSF-1R-dependent osteolysis in an experimental breast cancer bone metastasis model. Clin Exp Metastasis 20 (8): 757-66, 2003.
- Wagner AJ, Morgan JA, Chugh R, et al.: Inhibition of heat shock protein 90 (Hsp90) with the novel agent IPI-504 in metastatic GIST following failure of tyrosine kinase inhibitors (TKIs) or other sarcomas: clinical results from phase I trial. [Abstract] J Clin Oncol 26 (suppl 15): A-10503, 2008.
- Singer S, Rubin BP, Lux ML, et al.: Prognostic value of KIT mutation type, mitotic activity, and histologic subtype in gastrointestinal stromal tumors. J Clin Oncol 20 (18): 3898-905, 2002.
- Kim TW, Lee H, Kang YK, et al.: Prognostic significance of c-kit mutation in localized gastrointestinal stromal tumors. Clin Cancer Res 10 (9): 3076-81, 2004.
- Andersson J, Bümming P, Meis-Kindblom JM, et al.: Gastrointestinal stromal tumors with KIT exon 11 deletions are associated with poor prognosis. Gastroenterology 130 (6): 1573-81, 2006.
- Antonescu CR: Targeted therapy of cancer: new roles for pathologists in identifying GISTs and other sarcomas. Mod Pathol 21 (Suppl 2): S31-6, 2008.
- Debiec-Rychter M, Dumez H, Judson I, et al.: Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer 40 (5): 689-95, 2004.
- Heinrich MC, Corless CL, Demetri GD, et al.: Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 21 (23): 4342-9, 2003.
- Heinrich MC, Shoemaker JS, Corless CL, et al.: Correlation of target kinase genotype with clinical activity of imatinib mesylate (IM) in patients with metastatic GI stromal tumors (GISTs) expressing KIT (KIT+). [Abstract] J Clin Oncol 23 (Suppl 16): A-7, 3s, 2005.
- Dagher R, Cohen M, Williams G, et al.: Approval summary: imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res 8 (10): 3034-8, 2002.
- van Oosterom AT, Judson I, Verweij J, et al.: Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet 358 (9291): 1421-3, 2001.
- Wolter P, Stefan C, Decallonne B, et al.: The clinical implications of sunitinib-induced hypothyroidism: a prospective evaluation. Br J Cancer 99 (3): 448-54, 2008.
- Chu TF, Rupnick MA, Kerkela R, et al.: Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 370 (9604): 2011-9, 2007.
- Frye RF, Fitzgerald SM, Lagattuta TF, et al.: Effect of St John’s wort on imatinib mesylate pharmacokinetics. Clin Pharmacol Ther 76 (4): 323-9, 2004.
- Dutreix C, Peng B, Mehring G, et al.: Pharmacokinetic interaction between ketoconazole and imatinib mesylate (Glivec) in healthy subjects. Cancer Chemother Pharmacol 54 (4): 290-4, 2004.
- de Groot JW, Zonnenberg BA, Plukker JT, et al.: Imatinib induces hypothyroidism in patients receiving levothyroxine. Clin Pharmacol Ther 78 (4): 433-8, 2005.
- Bolton AE, Peng B, Hubert M, et al.: Effect of rifampicin on the pharmacokinetics of imatinib mesylate (Gleevec, STI571) in healthy subjects. Cancer Chemother Pharmacol 53 (2): 102-6, 2004.
- O’Brien SG, Meinhardt P, Bond E, et al.: Effects of imatinib mesylate (STI571, Glivec) on the pharmacokinetics of simvastatin, a cytochrome p450 3A4 substrate, in patients with chronic myeloid leukaemia. Br J Cancer 89 (10): 1855-9, 2003.
Resectable Primary GIST
General principles for the surgical therapy of gastrointestinal stromal tumors (GIST) include the following:
- All GIST 2 cm or larger in size are typically resected; the management of incidentally encountered GIST smaller than 2 cm in size remains controversial. There is no evidence that patients should undergo re-excision in cases in which there is complete resection of all macroscopic disease but microscopically margins are positive; watchful waiting and adjuvant imatinib may be appropriate for these patients.[1,2] In general, gastric GIST 5 cm or smaller in size may be removed by laparoscopic wedge resection. Because GIST rarely involve the locoregional lymph nodes, extensive lymph node resection or resection is rarely indicated. These tumors may have fragile pseudocapsules, so care must be taken to avoid rupturing the pseudocapsule during surgery, which could result in peritoneal dissemination.
- Therapy with postoperative adjuvant imatinib for GIST patients with completely resected localized disease is under investigation. This is a very heterogeneous population in terms of risk of relapse and death after surgical resection (see Table 1). Depending on mitotic count, tumor size, and tumor site, the risk of relapse after complete gross resection may be considerable. Results from two trials suggest that adjuvant imatinib reduces recurrence after complete resection of localized, primary GIST.[3,4] However, it is not clear whether improvements in recurrence with adjuvant therapy will translate into improved survival. In addition, the optimal duration of adjuvant imatinib is unknown.
In a single-arm, open-label, phase II multicenter study (ACOSOG-Z9000), patients underwent complete gross resection of KIT-expressing primary GIST that were at high risk of recurrence (tumor size >10 cm, tumor rupture, or <5 peritoneal metastases) and received a daily dose of imatinib for 1 year. One hundred and seven patients with gastric (50%) and intestinal (42%) GIST and a median tumor size of 13 cm were evaluable with a median follow-up of 4 years. The 1-, 2- and 3-year rates for overall survival (OS) were 99%, 97%, and 97%, respectively, while the 1-, 2- and 3-year recurrence-free survival (RFS) rates were 94%, 73%, and 61%, respectively.[4][Level of evidence: 3iiiDiii]
In one randomized, double-blinded phase III trial (ACOSOG-Z9001), 713 patients who had undergone complete gross resection of a primary GIST measuring at least 3 cm and expressing KIT had been treated with 1 year of imatinib (400 mg daily) or placebo.[3] The original primary endpoint of the trial was OS. However, when the overall death rate from GIST was shown to be low because of the efficacy of imatinib for advanced or recurrent disease, the investigators changed the endpoint to RFS. The trial was subsequently stopped early because of an interim analysis, which showed that patients who had been assigned to the imatinib arm experienced a 1-year RFS of 98%, whereas those assigned to the placebo arm had a 1-year RFS of 83% (overall HR = 0.35; 95% confidence interval (CI), 0.22–0.53; P < .0001). Since quality of life (QoL) was not measured in the study, it was not clear whether delayed RFS translated into improved patient QoL. No difference in OS was observed between the two arms, possibly because of the fairly short follow-up time and the crossover design of the study.[3][Level of evidence: 1iDii] In the course of the trial, at 48 months, there had been 5 deaths of the original 359 patients in the imatinib arm (about 1%) and 8 deaths of the original 354 patients in the placebo arm (about 2%) (P = .47). It is possible that imatinib was simply delaying recurrences that could have been treated equally well at relapse in those patients who had a recurrence of their tumors. In fact, the RFS curves appeared to converge after about 30 months of follow-up. The question of whether RFS will translate into improved OS may be answered by an ongoing Intergroup trial (EORTC-62024) in which patients are randomly assigned to 2 years of adjuvant imatinib versus observation, with OS as the primary endpoint.
Given the current uncertainties about the overall impact of adjuvant imatinib, shared decision making with the patient is important. Since the cost of administration of imatinib for a year or more may be high, knowledge about what the patient may have to pay out of pocket may help inform the discussion.[5]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Demetri GD, Benjamin RS, Blanke CD, et al.: NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST)–update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw 5 (Suppl 2): S1-29; quiz S30, 2007.
- Otani Y, Furukawa T, Yoshida M, et al.: Operative indications for relatively small (2-5 cm) gastrointestinal stromal tumor of the stomach based on analysis of 60 operated cases. Surgery 139 (4): 484-92, 2006.
- Dematteo RP, Ballman KV, Antonescu CR, et al.: Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet 373 (9669): 1097-104, 2009.
- DeMatteo RP, Owzar K, Antonescu CR, et al.: Efficacy of adjuvant imatinib mesylate following complete resection of localized, primary gastrointestinal stromal tumor (GIST) at high risk of recurrence: the U.S. Intergroup phase II trial ACOSOG Z9000. [Abstract] American Society of Clinical Oncology 2008 Gastrointestinal Cancers Symposium, 25-27 January 2008, Orlando, FL. A-8, 2008.
- Kelley RK, Venook AP: Nonadherence to imatinib during an economic downturn. N Engl J Med 363 (6): 596-8, 2010.
Unresectable Primary GIST
Therapy with neoadjuvant imatinib is under evaluation. It may be used for patients with very large primary gastrointestinal stromal tumors (GIST) or poorly positioned small GIST (considered unresectable without the risk of unacceptable morbidity or functional deficit) until surgical therapy is feasible, which can take as long as 6 to 12 months.[1,2] There are no controlled trials addressing the benefits of imatinib in this setting, so the impact of neoadjuvant imatinib on overall survival (OS) is unclear. Even the conversion rate from inoperability to operability with neoadjuvant therapy is ill-defined. Therefore, any advantages of neoadjuvant therapy are currently theoretical.
Mutational analysis may help to exclude nonsensitive mutations prior to imatinib cytoreduction therapy or to determine whether a tumor harbors a KIT exon 9 mutation, potentially requiring an increase in initial imatinib dosing.[1,3] Follow-up is performed at close intervals, possibly with fluorine F 18-fludeoxyglucose positron emission tomography (18F-FDG PET), to assess response to therapy.[1,4]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Demetri GD, Benjamin RS, Blanke CD, et al.: NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST)–update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw 5 (Suppl 2): S1-29; quiz S30, 2007.
- Bonvalot S, Eldweny H, Péchoux CL, et al.: Impact of surgery on advanced gastrointestinal stromal tumors (GIST) in the imatinib era. Ann Surg Oncol 13 (12): 1596-603, 2006.
- Debiec-Rychter M, Sciot R, Le Cesne A, et al.: KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 42 (8): 1093-103, 2006.
- Gayed I, Vu T, Iyer R, et al.: The role of 18F-FDG PET in staging and early prediction of response to therapy of recurrent gastrointestinal stromal tumors. J Nucl Med 45 (1): 17-21, 2004.
Metastatic or Recurrent GIST
The primary treatment of patients with metastatic or recurrent gastrointestinal stromal tumors (GIST) involves medical therapy with a tyrosine kinase inhibitor (TKI); in select cases, surgical therapy may be added. Patients with metastatic or recurrent tumors that do not respond to these measures may be candidates for clinical trials.
- Therapy with imatinib is standard for patients with metastatic or recurrent disease. The initial dose may range from 400 mg to 600 mg daily, except for patients with tumors containing KIT exon 9 mutations, who may receive 800 mg daily.[1] Response is evaluated with computed tomography (CT), magnetic resonance imaging (MRI), or fluorine F 18-fludeoxyglucose positron emission tomography (18F-FDG PET).[2,3,4,5,6] Treatment is usually continued indefinitely in the absence of disease progression or unacceptable toxicity.[3,7]
In a phase III randomized trial involving 746 patients with advanced incurable GIST , higher dose treatment with 800 mg imatinib daily did not show any advantage over lower-dose treatment with 400 mg imatinib daily as primary systemic therapy; no statistically significant differences in objective response rates, progression-free survival (PFS), or overall survival (OS) were observed.[8][Levels of evidence: 1iiA; 1iiDiii; and 1iiDiv].
However, in a phase II randomized trial examining dose selection in 946 patients with advanced GIST (EORTC-62005 [NCT00685828I]), pretreatment samples of GIST from 377 patients were analyzed for KIT gene mutations; those patients whose tumors expressed an exon 9 KIT mutation and were treated with a daily dose of 800 mg of imatinib (vs. 400 mg) experienced a significantly superior PFS (P = .0013) with a reduction of relative risk of 61%.[1][Level of evidence: 1iiDiii]
In the event of progression of tumors without KIT exon 9 mutations on lower dose imatinib (i.e., 400 mg –600 mg daily), the imatinib dose may be increased to 800 mg daily (in split doses). Alternatively, in the management of imatinib resistance, the patient may be switched directly to sunitinib from low-dose imatinib.[9]
- In case of tumor progression or intolerance to imatinib, the second-line standard therapy consists of sunitinib administered at a dose of 50 mg daily in a 4-weeks-on/2-weeks-off regimen. Alternatively, a regimen consisting of a daily dose of 37.5 mg may be used.[10] As with imatinib therapy, the response to therapy with sunitinib is evaluated with CT, MRI, or 18F-FDG PET, and treatment is usually continued indefinitely in the absence of disease progression or unacceptable toxicity.[2,3,11]
In a randomized controlled trial involving 312 patients with imatinib-resistant GIST, median time to tumor progression was more than four times as long with sunitinib (27.3 weeks, 95% confidence interval (CI) 16.0–32.1) than with placebo treatment (6.4 weeks, 95% CI, 4.4–10.0; hazard ratio (HR) = 0.33; 95% CI, 0.23–0.47; P < .0001) on the basis of radiologic assessment. Overall survival was similarly better for sunitinib-treated patients (HR for death = 0.49; 95% CI, 0.29–0.83).[10][Level of evidence: 1iA]
It has been suggested that patients with limited disease progression continue on imatinib or sunitinib at a dose that can be tolerated. For patients with large bulky tumors who are receiving imatinib, there may be a 5% risk of tumor hemorrhage unassociated with thrombocytopenia;[12,13] accordingly, patients with large, high-risk tumors are monitored very closely for evidence of a decline in hemoglobin levels during the first 4 to 8 weeks of imatinib therapy. All patients on TKI therapy are closely monitored for tumor response and side effects, which may require dose reductions, interruptions, or cessation of TKI therapy in cases of persistent, excessive toxicity. In addition, dose modification of the TKI or substitution with medications that do not affect CYP450 3A4 levels may be necessary for patients taking drugs that affect CYP450 3A4 levels.[3]
- Surgery may be added to medical therapy for selected patients with GIST in an effort to delay or prevent recurrence, although the benefit of this therapeutic approach in metastatic GIST has yet to be proven in a randomized clinical trial.
In a retrospective study involving 69 consecutive patients who underwent surgery for unresectable primary or metastatic GIST while receiving kinase inhibitors, patients with stable disease or limited progression were found to have prolonged survival after debulking procedures.[14] In this group of GIST patients, no evidence of disease was found in 78%, 25%, and 7% of patients with stable disease, limited progression, and generalized progression, respectively, after surgery; the 12-month PFS was 80%, 33%, and 0% for patients with stable disease, limited progression, and generalized progression, whereas the 12-month OS was 95%, 86%, and 0% for the same patient subsets.[14][Levels of evidence: 3iiDii and 3iiA] The authors of this study concluded that surgery for patients with generalized progression be limited to a palliative role.
Overall, the indications for surgery in the management of metastatic or recurrent GIST include the following:[3]
- For stable disease (i.e. disease that is stable or shrinking on TKI therapy when gross resection is possible).
- For limited disease progression (i.e., isolated tumor deposits that are progressing on TKI therapy after initial response [indicating delayed drug resistance], while other sites of disease remain stable).
- For oncologic emergencies including hemorrhage, perforation, obstruction, or abscess.
The first two indications (1 and 2) identify subsets of patients with advanced disease that are selected for relative disease stability. Therefore, the favorable outcomes that have been noted in case series may be principally the result of selection bias rather than true benefit from surgery. Because the median time to the development of secondary resistance to imatinib has been found to be about 2 years,[15] it has been suggested that surgery for metastatic or recurrent disease in patients on imatinib/sunitinib be performed before 2 years. Most experts would recommend considering surgery after 6 to 12 months of disease stability with TKI therapy.[3] Drug therapy may be continued after surgery. Patients with generalized disease progression are managed medically and may be enrolled in clinical trials.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Debiec-Rychter M, Sciot R, Le Cesne A, et al.: KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 42 (8): 1093-103, 2006.
- Demetri GD: Gastrointestinal stromal tumor. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1060-73.
- Demetri GD, Benjamin RS, Blanke CD, et al.: NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST)–update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw 5 (Suppl 2): S1-29; quiz S30, 2007.
- Blanke CD, von Mehren M, Joensuu H, et al.: Evaluation of the safety and efficacy of an oral molecularly-targeted therapy, STI157, in patients (pts) with unresectable or metastatic gastrointestinal stromal tumors (GISTs) expressing c-kit (CD117). [Abstract] Proceedings of the American Society of Clinical Oncology 20: A-1, 1a, 2001.
- van Oosterom AT, Judson I, Verweij J, et al.: Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet 358 (9291): 1421-3, 2001.
- Choi H, Charnsangavej C, Faria SC, et al.: Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol 25 (13): 1753-9, 2007.
- Blay JY, Le Cesne A, Ray-Coquard I, et al.: Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: the French Sarcoma Group. J Clin Oncol 25 (9): 1107-13, 2007.
- Blanke CD, Rankin C, Demetri GD, et al.: Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol 26 (4): 626-32, 2008.
- Rutkowski P, Nowecki Z, Nyckowski P, et al.: Surgical treatment of patients with initially inoperable and/or metastatic gastrointestinal stromal tumors (GIST) during therapy with imatinib mesylate. J Surg Oncol 93 (4): 304-11, 2006.
- Demetri GD, van Oosterom AT, Garrett CR, et al.: Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 368 (9544): 1329-38, 2006.
- Prior JO, Montemurro M, Orcurto MV, et al.: Early prediction of response to sunitinib after imatinib failure by 18F-fluorodeoxyglucose positron emission tomography in patients with gastrointestinal stromal tumor. J Clin Oncol 27 (3): 439-45, 2009.
- Demetri GD, von Mehren M, Blanke CD, et al.: Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 347 (7): 472-80, 2002.
- Dagher R, Cohen M, Williams G, et al.: Approval summary: imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res 8 (10): 3034-8, 2002.
- Raut CP, Posner M, Desai J, et al.: Surgical management of advanced gastrointestinal stromal tumors after treatment with targeted systemic therapy using kinase inhibitors. J Clin Oncol 24 (15): 2325-31, 2006.
- Verweij J, Casali PG, Zalcberg J, et al.: Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet 364 (9440): 1127-34, 2004.
Resistant / Refractory GIST
Eventual development of resistance to both imatinib and sunitinib is nearly universal. There is no standard therapy when this occurs, and patients should be considered for investigational therapy. Additional oral TKIs are under investigation. For example, in a preliminary case series, nilotinib has shown biological activity, with unknown impact on survival.[1]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Montemurro M, Schöffski P, Reichardt P, et al.: Nilotinib in the treatment of advanced gastrointestinal stromal tumours resistant to both imatinib and sunitinib. Eur J Cancer 45 (13): 2293-7, 2009.
Changes to This Summary (02 / 07 / 2018)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Stage Information for Gastrointestinal Stromal Tumors
Editorial changes were made to this section.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® – NCI’s Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of gastrointestinal stromal tumors. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewer for Gastrointestinal Stromal Tumors Treatment is:
- Russell S. Berman, MD (New York University School of Medicine)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website’s Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Gastrointestinal Stromal Tumors Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/soft-tissue-sarcoma/hp/gist-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389157]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Last Revised: 2018-02-07