Sickle Cell Disease
Topic Overview
What is sickle cell disease?
Sickle cell disease changes normal, round red blood cells into cells that can be shaped like crescent moons. The name “sickle cell” comes from the crescent shape of the cells. (A sickle is a tool with a crescent-shaped blade.)
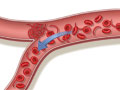
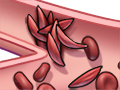
Normal red blood cells move easily through your blood vessels, taking oxygen to every part of your body. But sickled cells can get stuck and block blood vessels, which stops the oxygen from getting through. That can cause a lot of pain. It can also harm organs, muscles, and bones.
See a picture of sickle cells blocking a blood vessel.
Having sickle cell disease means a lifelong battle against the health problems it can cause, such as pain, infections, anemia, and stroke. But many people are able to have a very good quality of life by learning to manage the disease.
What causes sickle cell disease?
Sickle cell disease is inherited, which means it is passed from parent to child. To get sickle cell disease, a child has to inherit two sickle cell genes—one from each parent.
When a child inherits the gene from just one parent, that child has sickle cell trait. Having this trait means that you don’t have the disease but you are a carrier and could pass the gene on to your children.
What are the symptoms?
Painful events (sickle cell crises) are the most common symptom of sickle cell disease. They are periods of pain that happen when sickled cells get stuck in blood vessels and block the blood flow. These events usually cause pain in the hands, feet, belly, back, or chest. The pain may last for hours or for days.
People with sickle cell disease often have anemia, caused by a shortage of red blood cells. Anemia makes you feel weak and tired. People with sickle cell anemia may look pale or washed out. Their skin and the whites of their eyes may have a yellowish look (jaundice).
How is sickle cell disease diagnosed?
A simple blood test can show whether a person has sickle cell disease. Most states test for sickle cell disease before infants go home from the hospital.
How is it treated?
Self-care and medical treatment can help you manage pain and avoid other health problems.
Early treatment includes daily antibiotics from 2 months to 5 years of age to help prevent infections. Routine childhood and adult immunizations are also important.
Managing pain is often a big part of having sickle cell disease. You can prepare for a painful event ahead of time by creating a pain management plan with your doctor. The plan should include what you can do at home to relieve pain for yourself or your child. The plan should also tell you when it is best to call a doctor or go to a hospital.
Some people need regular blood transfusions to lower the risk of stroke and treat anemia and other problems. Some people take medicine to prevent complications. In rare cases, a stem cell transplant might be an option.
Regular checkups are an important part of life with this disease. People with sickle cell disease need a good working relationship with a doctor who is an expert in treating it.
How do you manage life with sickle cell disease?
Tips for you
- Learn what triggers, or sets off, painful events called sickle cell crises. Triggers often include cold temperatures, wind, dehydration, and too much exercise. Low oxygen caused by cigarette smoke, high altitude, and plane flights is another common trigger.
- People with sickle cell disease and their families face ongoing stress. A support network can help ease stress and worry. Ask your doctor if there is a support group in your area.
Tips for helping your child
- Make sure that your child takes antibiotics regularly until age 5 to prevent infections. And make sure he or she receives all the usual immunizations on schedule.
- Your child can take part in normal school activities. Make sure that teachers understand your child’s special needs, like needing frequent drinks and bathroom trips and avoiding overexertion and cold temperatures.
Cause
Sickle cell disease is an inherited blood disorder, passed from parent to child. Children with sickle cell disease have two defective hemoglobin S genes, one from each parent. Various forms of sickle cell disorder occur when a person inherits one hemoglobin S gene (sickle cell gene) from one parent and one other type of defective hemoglobin gene from the other parent.
Normally, a person inherits two genes that tell the body to produce normal hemoglobin A. One gene comes from each parent. People who inherit one defective hemoglobin S gene and one normal hemoglobin A gene have sickle cell trait. These people don’t have symptoms of sickle cell disease, and their bodies don’t make sickled blood cells. But they can pass the defective hemoglobin S gene to their children.
Symptoms
Painful events (sickle cell crises) in the hands or feet, belly, back, or chest are the most common symptom of sickle cell disease. This pain may last from hours to days. Most people with sickle cell disease also get anemia.
When a child is born with sickle cell disease, it isn’t possible to predict which symptoms will appear, when they will start, or how bad they will be.
Symptoms related to chronic anemia
Most people who have sickle cell disease have at least mild symptoms of chronic anemia, which may include:
- Weakness.
- Tiredness (fatigue).
- Pale appearance.
- Yellowing of the skin and the whites of the eyes (jaundice).
- Shortness of breath, especially when they are active.
Severe anemia may raise the chance of a person with sickle cell disease getting high blood pressure in the lungs (pulmonary hypertension). This can be deadly.
Symptoms caused by sickle cell crisis
Painful sickle cell crisis symptoms are caused by blocked blood vessels in bones, organs, and other tissues. This can cause extreme pain for hours or days. These painful events can occur rarely to often. Sometimes home treatment can help the pain. And sometimes a hospital stay is needed.
Infants and young children may have episodes of extreme pain in the hands, the feet, or both ( hand-foot syndrome).
What Happens
Normal red blood cells have a 120-day life span. But people born with sickle cell disease have sickle-shaped blood cells that usually live no more than 20 days. These sickled cells can get stuck in blood vessels, blocking blood flow.
Less blood flow can damage the body’s organs, muscles, and bones, sometimes leading to life-threatening conditions. Sickle cell disease may cause problems such as:
- Sickle cell crisis, which happens when blood vessels are blocked. This is a common condition of sickle cell disease.
- Splenic sequestration, usually seen in infants and young children during or after a simple respiratory infection. Large numbers of sickled red blood cells get trapped in the spleen. It can cause sudden and life-threatening anemia.
- Acute chest syndrome, most common in children but more severe in adults. Symptoms include coughing and chest pain, which may occur after an infection or painful event.
- Severe infections.
- Aplastic crisis, which may occur after infection with some viruses. During an aplastic crisis, bone marrow stops producing red blood cells, which causes sudden and severe anemia.
When a child is born with sickle cell disease, it’s impossible to predict which problems will develop, when they will start, or how bad they will be. During the first 6 months of life, infants have a high level of fetal hemoglobin (HbF) in their blood, which protects them from red blood cell sickling. But dangerous complications of sickle cell disease may quickly develop between ages 6 months and 5 years, after levels of fetal hemoglobin decrease.
Older children and adults with sickle cell disease may have few problems. Or they may have a pattern of ongoing complications that shortens their lives. The most common and serious problems caused by sickle cell disease are anemia, pain, and organ failure. Stroke affects around 10 out of 100 children who have sickle cell disease.footnote 1
Other complications of sickle cell disease include:
- Growth slowdown. Children who have sickle cell disease often grow more slowly than normal and go through puberty later than children who don’t have the disease.
- Open sores (ulcers) on the legs and feet, commonly during adulthood. These ulcers can be very painful and heal slowly. Some may last for years.
- Eye damage. Long-term vision problems may result from blocked blood flow in the inner lining of the eye (retina).
- A lung problem called pulmonary hypertension where blood pressure is very high in the lungs.
What Increases Your Risk
A child’s risk of getting sickle cell disorder occurs when he or she inherits one sickle cell gene and one other type of defective hemoglobin gene.
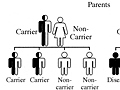
People who inherit one defective hemoglobin S gene and one normal hemoglobin A gene have sickle cell trait. They don’t have symptoms of sickle cell disease, and their bodies don’t make sickled blood cells. But they have a 1-out-of-2 (50%) chance of passing the defective hemoglobin S gene to each of their children.
- If both parents have sickle cell trait, each of their children will have a 1-out-of-4 (25%) chance of having sickle cell disease.
- If one parent has sickle cell disease (has two genes for making hemoglobin S) and the other has sickle cell trait (has one hemoglobin S gene and one normal hemoglobin A gene), each of their children will have a 1-out-of-2 (50%) chance of having sickle cell disease and a 1-out-of-2 (50%) chance of having sickle cell trait.
- If one parent has sickle cell disease (two hemoglobin S genes) and the other has two normal hemoglobin A genes, each of their children will have sickle cell trait. None of the children will have sickle cell disease.
People whose ancestors were from Africa, India, the Middle East, the Mediterranean (Turkey, Italy, Greece), and some Latin American countries are more likely to inherit the gene that can cause sickle cell disease. In the United States, about 2,000 children are born each year with sickle cell disease.footnote 2
For more information, see a picture of the risk of passing on an autosomal recessive disease such as sickle cell disease.
When should you call your doctor?
Call 911 or other emergency services immediately if you have sickle cell disease and one or more of the following symptoms are present:
- Difficulty breathing or shortness of breath
- Chest pain
- Severe abdominal (belly) pain
- Sudden weakness
- Sudden numbness or tingling in the hands, feet, fingers, or toes (even if it goes away on its own)
- Sudden poor balance and poor coordination when walking (even if it goes away on its own)
- Confusion (even if it goes away on its own)
- Garbled speech or an inability to speak (even if it goes away on its own)
- Sudden change in vision
- Severe headache
- Loss of consciousness
- Fever higher than 101°F (38.33°C)
- Severe cough
- Repeated vomiting or persistent diarrhea
- A sudden increase in the size of your or your child’s spleen. (Learn from your doctor how to feel your child’s spleen to check its size.)
- Increased paleness
- Lightheadedness
- Persistent erection of the penis (priapism) that lasts more than 2 to 3 hours or is extremely painful
- Severe pain that can’t be relieved with your usual prescription painkilling drugs or other pain-relief methods
Call your doctor if you or your child has any of the following symptoms:
- A painful event (sickle cell crisis)
- An open sore (ulcer) on the leg
- More frequent urination than usual
Make a pain management plan with your doctor that includes where and when to get treatment in case of a sickle cell emergency.
Painful events (crises) can be treated at home depending on how severe the pain is and how long you’ve had it. For more information, see Home Treatment.
Who to see
If you or your child has sickle cell disease, try to find a doctor who has special training for this disease. Some medical centers and hospitals specialize in sickle cell disease treatment and support. If your local community doesn’t offer this option, look for a doctor and a pain treatment specialist who have experience in treating sickle cell disorders. Choose a doctor you are comfortable with and can partner with over the long term.
The following types of health professionals can diagnose and help treat symptoms of sickle cell disease. Some of these health professionals may provide specialized treatment or counseling.
Exams and Tests
Sickle cell disease is diagnosed when initial tests show abnormal hemoglobin, with more testing if needed. A sickle cell test looks for sickle cell trait and sickle cell disease.
Prenatal testing
Doctors can diagnose sickle cell disease before a child is born (prenatally). Couples who are at risk for passing on this disease to their children may want to talk with a genetic counselor about prenatal testing.
Prenatal tests include:
Infant screening
Sickle cell disease can be diagnosed at birth. Most states in the United States screen all newborns for sickle cell disease along with other common disorders. You can also ask for screening.
Soon after birth, a sample of blood is taken from the infant’s heel and sent to a lab, where it is screened for the presence of sickle cell hemoglobin (hemoglobin S).
Adult screening
If one member of a couple has sickle cell disease or sickle cell trait, the other member should be tested before becoming pregnant. This test requires a blood sample, which is screened for the presence of hemoglobin S, hemoglobin C, or beta-thalassemia.
If one or both members of a couple carry a hemoglobin S gene or another abnormal hemoglobin gene, the couple may want to meet with a genetic counselor before becoming pregnant to learn more about their chances of having a child with sickle cell disease. Your doctor can help you find a genetic counselor to discuss a genetic test.
Pulmonary hypertension is a severe, common problem for people who have sickle cell disease. It can be detected early with an echocardiogram, a painless method of measuring blood flow.
Treatment Overview
Treatment involves getting routine tests to monitor health, managing pain events (crises), and treating related health problems as they arise.
Treatment for severe cases of sickle cell disease may include medicines. For more information, see Medications.
Treatment for children
When parents learn that their baby has sickle cell disease, it’s the beginning of a lifelong education process. Knowing as much as you can about the disease can help you control symptoms as they arise and know what to do in emergency situations. Treatment includes:
- Routine childhood immunizations. Immunizations in adulthood are important too.
- Daily antibiotics from 2 months to 5 years of age to prevent life-threatening infections. This practice stops at age 5 because older children don’t have as many severe infections.
- The medicine hydroxyurea.
- Multivitamin supplements with iron during infancy.
- Folic acid supplements daily.
- Protein supplements if there is a lag in weight gain.
Starting at age 2 years, your child should get screened every now and then with a transcranial ultrasound. This test measures blood flow in the arteries of the head and neck. If test results show a high chance for stroke, your child may get blood transfusions to lower the risk.footnote 3
Tests to monitor treatment
Routine tests include:
- Complete blood count (CBC).
- Urine test.
- Tests to monitor the functioning of organs.
- Tests to check for vision problems.
- Tests for Hepatitis C infection if frequent blood transfusions are given.
Managing pain
Pain is sometimes a chronic problem for people with sickle cell disease. Your doctor or a pain treatment specialist can help you develop pain management skills. These skills include distraction, guided imagery, deep breathing, relaxation, and positive self-talk.
Painful events can happen suddenly and unpredictably and can become life-threatening. Bouts of severe pain can last for hours to days and are difficult to treat. They’re exhausting for caregivers as well as for the person in pain. For more information, see the topic Chronic Pain.
Severe episodes of prolonged erection of the penis (priapism) need evaluation by your doctor. Treatment may include fluids (hydration), pain medicines, treatment by a urologist, and blood transfusions.
There are also things you can do at home to manage pain. To learn more, see Home Treatment.
What to think about
A series of blood transfusions is the treatment of choice to prevent strokes and treat other aspects of sickle cell disease. Stem cell transplant is a rare treatment. For more information, see Other Treatment.
People with sickle cell disease should avoid contact with anyone suspected of having fifth disease, which is caused by parvovirus. Parvovirus can cause the body to temporarily stop making blood cells, a severe life-threatening problem in someone who has sickle cell disease. Aplastic anemia can occur as a result of a shortage of red blood cells. It can come on suddenly and is life-threatening if not treated.
People with sickle cell disease and their families face ongoing stress. For help coping, see Home Treatment.
Home Treatment
Home treatment for sickle cell disease includes steps to control pain and prevent complications of the disease. If you don’t already have a home treatment plan, ask your doctor to help you make one. Use this plan whenever symptoms are present. Your plan may include tips for:
- Managing pain.
- Home treatment for prolonged erection of the penis (priapism).
- Staying healthy.
- Coping with stress and worry.
Managing a child’s special needs
You can help your child cope with special needs in school by:
- Making arrangements with teachers or a tutor to help your child keep pace with classmates when illness causes absences from school.
- Explaining to teachers that children with sickle cell disease may need to use the bathroom more often than other kids. They also need more water than the other students. Not drinking enough water can raise the chance of a sickle cell crisis.
- Educating teachers and other school employees about the signs and symptoms of sickle cell disease that need urgent medical care. Written instructions will help school personnel know what to do and who to call in an emergency.
Children with sickle cell disease can usually exercise and play normally if they:
- Drink plenty of fluids before, during, and after exercise. Lack of fluids (dehydration) can cause cells to sickle.
- Get regular rest breaks during vigorous exercise.
- Stay warm. Exposure to cold air, wind, and water can trigger a sickle cell crisis. Dress children in warm layers of clothing for cold-weather activities. Your child should avoid swimming and playing in cold water.
Folic acid supplements are often a necessary part of the diet for people with sickle cell disease, particularly if you aren’t eating enough folate-rich leafy vegetables (such as spinach).
Medications
Medicines that treat sickle cell disease include hydroxyurea and various pain medicines. Some of these medicines require a prescription. Others are available over the counter. Pain medicine may work best when combined with pain management skills, such as distraction; guided imagery; deep breathing; relaxation; and positive, encouraging self-talk.
Pain treatment for sickle cell disease pain varies depending on how bad the pain is and how long the pain lasts. Medicines that treat the pain include over-the-counter pain relievers (such as ibuprofen) and prescription opioids (such as codeine). Opiate pain medicines are used under careful medical supervision.
Surgery
Some sickle cell complications are treated with surgery. Surgery options include:
- Removal of the spleen (splenectomy), to prevent the trapping of too many red blood cells in the spleen (splenic sequestration).
- Removal of the gallbladder (cholecystectomy), to prevent problems caused by gallstones.
- Draining fluid from the penis in cases of severe priapism.
- Hip replacement, if the tissue in the hip breaks down and dies because it doesn’t get enough blood (osteonecrosis).
Other Treatment
Blood transfusions may be used for sickle cell disease. They are the treatment of choice to prevent strokes and treat other aspects of this disease. They can reduce the risk of some complications and improve symptoms of severe anemia.
Stem cell transplants can cure sickle cell disease. But they are not common.
References
Citations
- Goldstein LB, et al. (2010). Guidelines for the primary prevention of stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. Published online December 2, 2010 (doi: 10.1161/STR.0b013e3181fcb238). Also available online: http://stroke.ahajournals.org/content/42/2/517.full.
- Wang WC (2009). Sickle cell anemia and other sickling syndromes. In JP Greer et al., eds., Wintrobe’s Clinical Hematology, 12th ed., pp. 1038–1082. Philadelphia: Lippincott Williams and Wilkins.
- Yawn BP, et al. (2014). Management of sickle cell disease: Summary of the 2014 evidence-based report by expert panel members. JAMA, 312(10): 1033–1048.
Current as of: March 28, 2019
Author: Healthwise Staff
Medical Review:E. Gregory Thompson, MD – Internal Medicine & Adam Husney, MD – Family Medicine & Martin J. Gabica, MD – Family Medicine & Martin H. Steinberg, MD – Hematology
This information does not replace the advice of a doctor. Healthwise, Incorporated, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the Terms of Use. Learn how we develop our content.